Placas, ovillos y una guerra casi religiosa
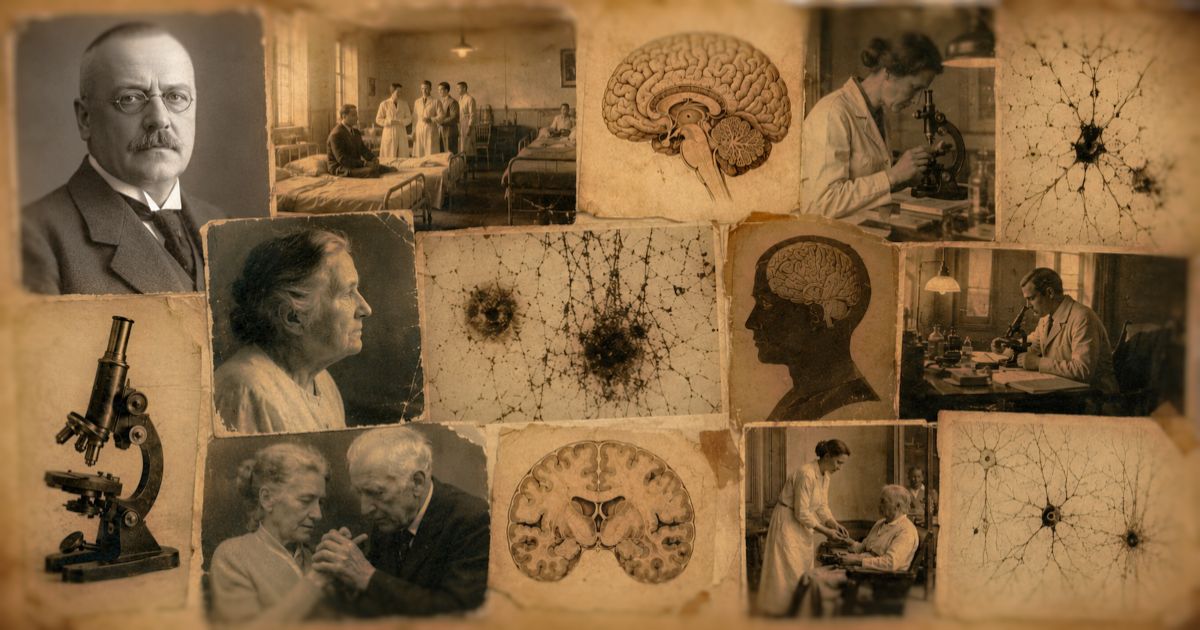
Durante el siglo XX, los neuropatólogos sabían que el Alzheimer tenía dos grandes marcas en el cerebro: placas seniles y ovillos neurofibrilares. Pero todavía faltaba conocer su composición.
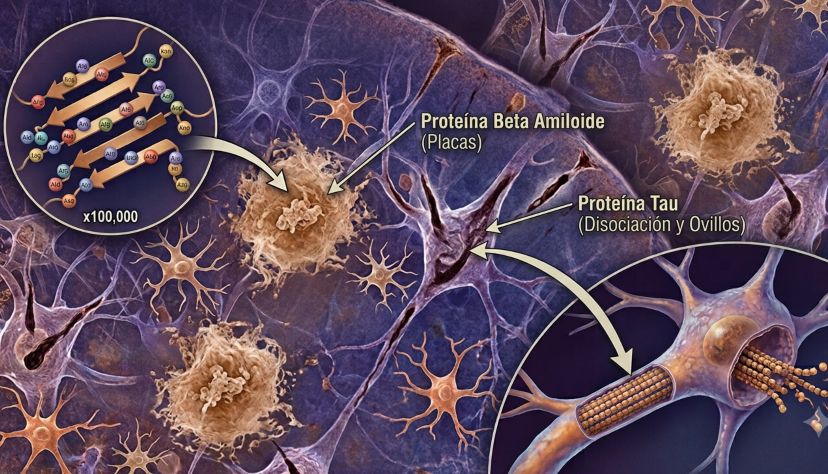
En los años ochenta se identificó que las placas estaban formadas por beta-amiloide, un pequeño fragmento de proteína que se acumula fuera de las neuronas. Poco después se demostró que los ovillos estaban compuestos por tau, una proteína que normalmente ayuda a estabilizar la estructura interna de la neurona, pero que en el Alzheimer se pliega mal y se enreda.
«La medicina había encontrado a sus dos grandes protagonistas: amiloide y tau.«
Entonces comenzó una discusión científica que, vista desde fuera, puede parecer extraña, pero que movió enormes cantidades de investigación, dinero y esperanza. ¿Cuál era el verdadero culpable? ¿El amiloide que se acumulaba como basura pegajosa entre las neuronas? ¿O la tau que se torcía dentro de ellas hasta asfixiarlas?
Algunos investigadores defendían la hipótesis amiloide: el depósito de beta-amiloide sería el evento inicial, la primera ficha del dominó. Luego vendría la alteración de tau, la muerte neuronal, la atrofia cerebral y, finalmente, la demencia. Otros insistían en que tau se relacionaba mucho mejor con los síntomas y con la gravedad de la enfermedad.
Durante años, los defensores del amiloide y los defensores de tau parecían bandos opuestos. El artículo reciente de JAMA lo describe con humor: los “baptists” del beta-amiloide contra los “Tauists”. Una especie de guerra santa de laboratorio.
«Pero, como ocurre muchas veces en medicina, la realidad terminó siendo más interesante que la pelea: ambos tenían parte de razón.»
El cromosoma 21 y la pista genética
La hipótesis amiloide ganó fuerza por una pista genética. La proteína precursora del amiloide, conocida como APP, se encuentra en el cromosoma 21. Eso era llamativo porque las personas con síndrome de Down tienen una copia extra de ese cromosoma y, con frecuencia, desarrollan cambios cerebrales tipo Alzheimer a edades tempranas.
Además, algunas familias con Alzheimer hereditario de inicio temprano tenían mutaciones relacionadas con APP o con proteínas que participan en su procesamiento, como las presenilinas. En esas familias, una sola copia alterada del gen podía condenar a varios miembros a desarrollar placas, ovillos, demencia y muerte prematura.
Parecía una prueba poderosa: si las mutaciones que aumentan el amiloide producen Alzheimer, entonces remover amiloide debería detener la enfermedad.
«La lógica era impecable. La realidad fue mucho más dura.«
La gran promesa que no llegó
Alrededor del año 2000, muchos científicos pensaban que la cura estaba cerca. Si el amiloide era el disparador, había que bloquearlo, retirarlo o impedir que se acumulara. Se probaron inhibidores de enzimas, vacunas y anticuerpos monoclonales diseñados para limpiar distintas formas de beta-amiloide.
Pero los resultados fueron decepcionantes. Algunos tratamientos empeoraban síntomas. Otros producían inflamación cerebral. Varios anticuerpos limpiaban poco, llegaban tarde o simplemente no mejoraban lo suficiente la evolución clínica. Medicamentos con nombres prometedores —bapineuzumab, solanezumab, gantenerumab, crenezumab— fueron cayendo uno tras otro en estudios fase 3.
Para 2020, después de dos décadas de investigación intensiva y miles de millones de dólares invertidos, incluso muchos defensores de la hipótesis amiloide comenzaron a dudar. ¿Habíamos perseguido al sospechoso equivocado? ¿Era el amiloide una causa, una consecuencia o sólo un acompañante?
La historia se volvió todavía más turbulenta con aducanumab.
Aducanumab: esperanza, controversia y desconfianza
Aducanumab fue un anticuerpo antiamiloide que prometió cambiar la historia. Sus estudios iniciales generaron entusiasmo, pero los ensayos clínicos grandes tuvieron resultados contradictorios. En 2019 fueron detenidos por falta de eficacia en un análisis intermedio. Después, al revisar más datos, uno de los estudios pareció mostrar beneficio y otro no.
La FDA aprobó aducanumab de manera acelerada en 2021, contra la recomendación de su propio comité asesor. La decisión provocó una tormenta: editoriales enfrentados, renuncias, investigaciones, críticas al proceso regulatorio y una pérdida importante de confianza. Para muchos médicos, aducanumab no fue la llegada triunfal del primer modificador de la enfermedad, sino una advertencia sobre cómo la desesperación terapéutica puede adelantar conclusiones.
Pero la historia no terminó ahí.
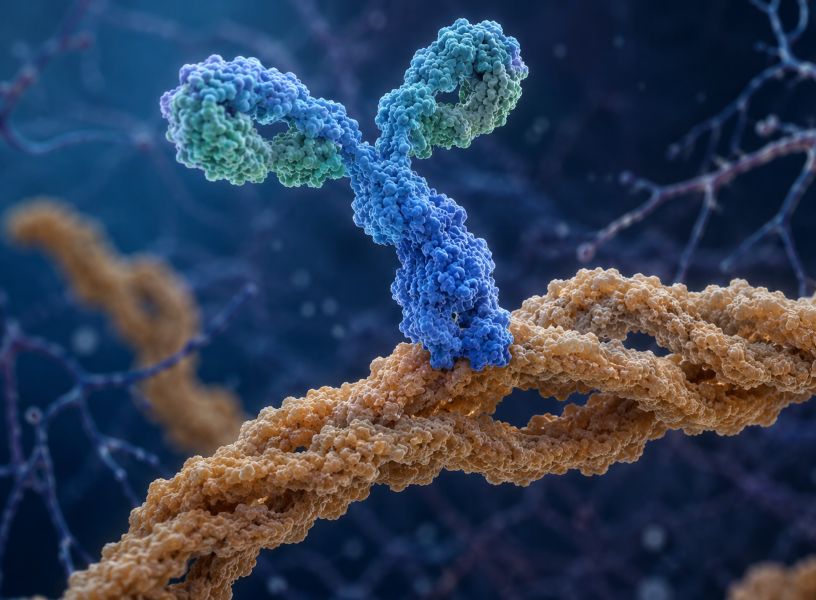
Lecanemab y donanemab: no curan, pero cambian el terreno
El campo volvió a moverse con lecanemab y donanemab, dos anticuerpos monoclonales antiamiloide estudiados en personas con Alzheimer temprano: deterioro cognitivo leve o demencia leve con biomarcadores positivos.
Estos tratamientos sí demostraron algo que durante décadas se había buscado: remover amiloide puede enlentecer la progresión clínica. No detiene la enfermedad. No devuelve la memoria perdida. No impide que el paciente continúe deteriorándose. Pero enlentece el declive aproximadamente un 30% en ensayos de 18 meses.
Eso, traducido a la vida cotidiana, puede ser modesto. Puede significar algunos meses de diferencia en la progresión. Para algunos, es poco. Para otros, especialmente familias que han visto perderse a alguien en la niebla del Alzheimer, unos meses de mayor autonomía pueden tener un valor enorme.
Sin embargo, estos tratamientos no son inocuos. Pueden producir ARIA, siglas en inglés de anomalías relacionadas con amiloide en imagen: edema cerebral, microhemorragias y, en casos raros, hemorragias graves o muerte. Por eso requieren selección cuidadosa, resonancias seriadas, discusión franca de riesgos y beneficios, y una medicina más precisa que entusiasta.
«El mensaje es claro: por primera vez tenemos tratamientos que modifican algo del curso biológico del Alzheimer, pero seguimos lejos de una cura.«
Los biomarcadores: cuando el Alzheimer dejó de ser sólo una sospecha
Uno de los grandes avances no fue un medicamento, sino la posibilidad de ver la enfermedad en vida.
Durante casi todo el siglo XX, el diagnóstico de Alzheimer era clínico y probabilístico. El médico podía sospecharlo por la historia, la exploración y las pruebas cognitivas, pero la confirmación definitiva llegaba después de la muerte, al examinar el cerebro.
Eso cambió con los biomarcadores: PET de amiloide, líquido cefalorraquídeo, PET de tau y, más recientemente, biomarcadores en sangre como p-tau217. Gracias a ellos, el Alzheimer pasó de ser una sospecha clínica a una enfermedad biológica medible.
Este cambio resolvió un problema enorme. Muchos ensayos antiguos incluían pacientes diagnosticados clínicamente como Alzheimer, pero algunos no tenían amiloide cerebral. Es decir, no tenían la enfermedad biológica que el medicamento intentaba tratar. Era como probar un antibiótico contra neumonía en un grupo donde una tercera parte en realidad no tenía infección bacteriana.
Los biomarcadores permitieron seleccionar mejor a los pacientes. Y eso ayudó a que los nuevos ensayos fueran más claros.
Pero también abrieron un dilema…
¿Se puede tener Alzheimer sin síntomas? (ver parte 3)

Autor: Juan Pablo Ledesma Heyer. Médico Internista y Geriatra. Conferencista, autor y divulgador de temas relacionados al envejecimiento desde un enfoque integral y humanista. Fundador del proyecto EnVejezSer
